-
The photophysics of salicylic acid derivatives in aqueous solution
I.P. Pozdnyakov, A. Pigliucci, N. Tkachenko, V.F. Plyusnin, E. Vauthey and H. Lemmetyinen
Journal of Physical Organic Chemistry, 22 (5) (2009), p449-454


DOI:10.1002/poc.1489 | unige:3174 | Abstract | Article PDF
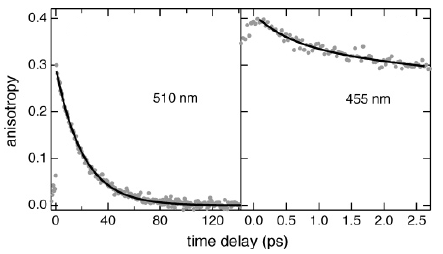
The photophysics of a number salicylic acid derivatives (SADs) in aqueous solutions was investigated in a wide range of pH by time-correlated single photon counting (λex = 350 nm, Ïresp = 300 ps) and fluorescence up-conversion (λex = 266 nm, Ïresp = 300 fs) techniques. The acid-base equilibrium constants in the ground (pKa) and the excited states (pKa*), the fluorescence quantum yields as well as the lifetimes of anionic, neutral, and cationic forms of SADs were determined. Evidence of ultrafast excited-state intramolecular proton transfer (ESIPT) leading to the formation of the proton-transferred excited state of SADs was obtained from the fluorescence up-conversion measurement. The nature of the ESIPT process is discussed.